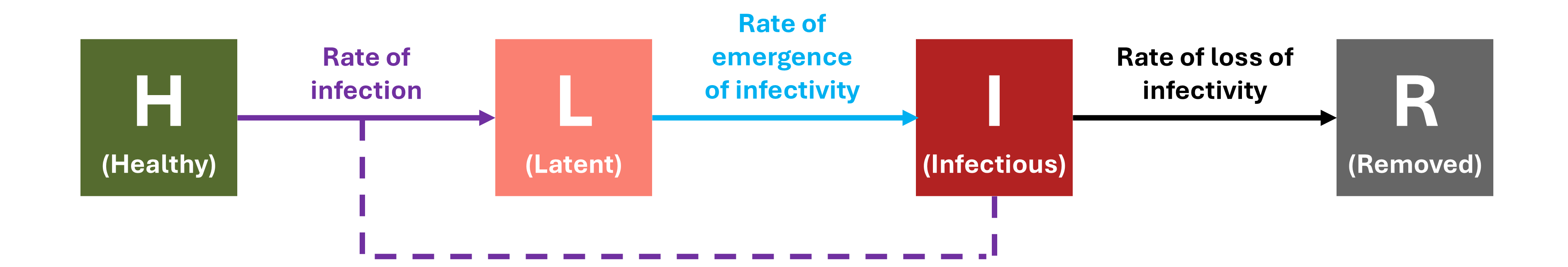
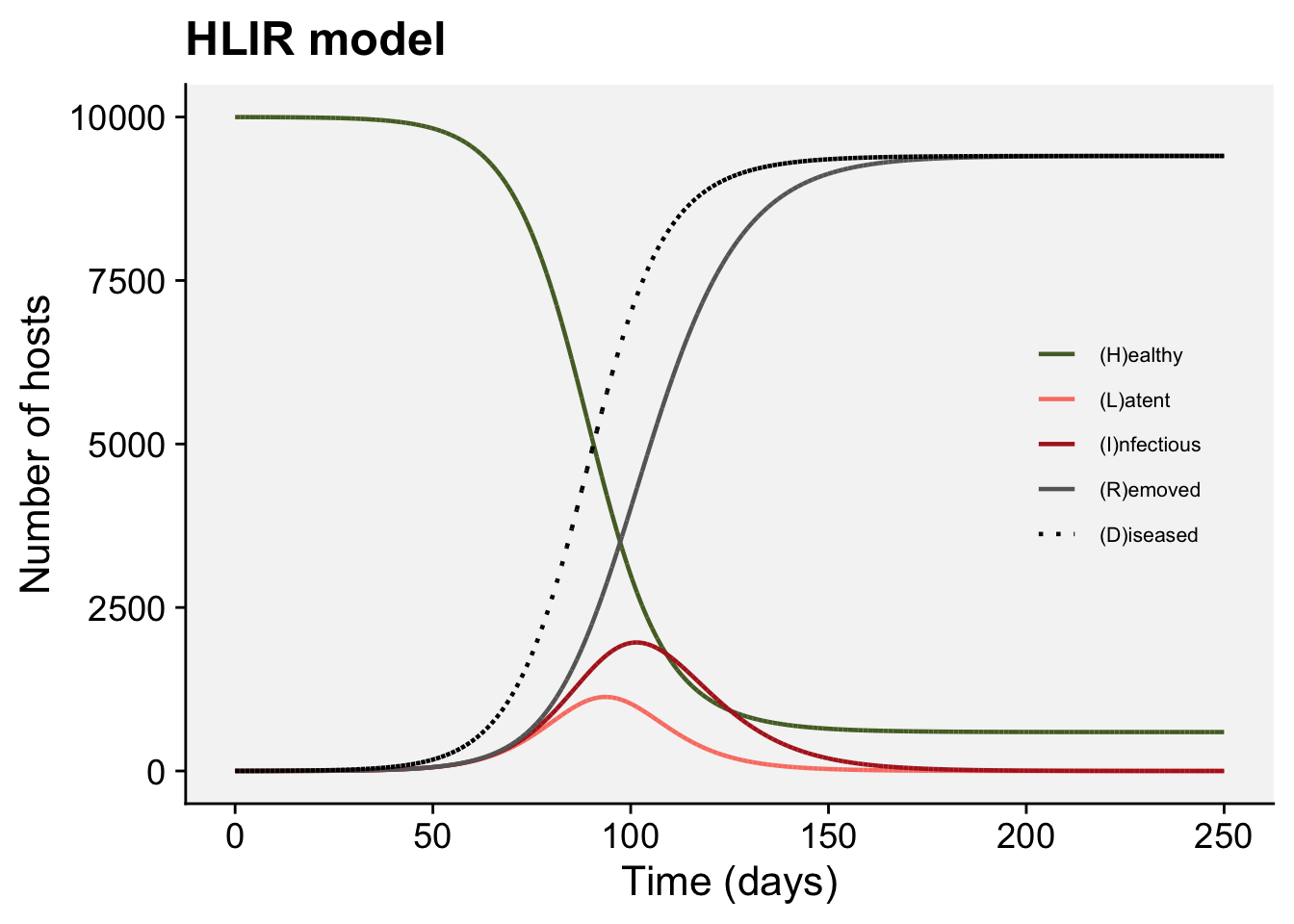
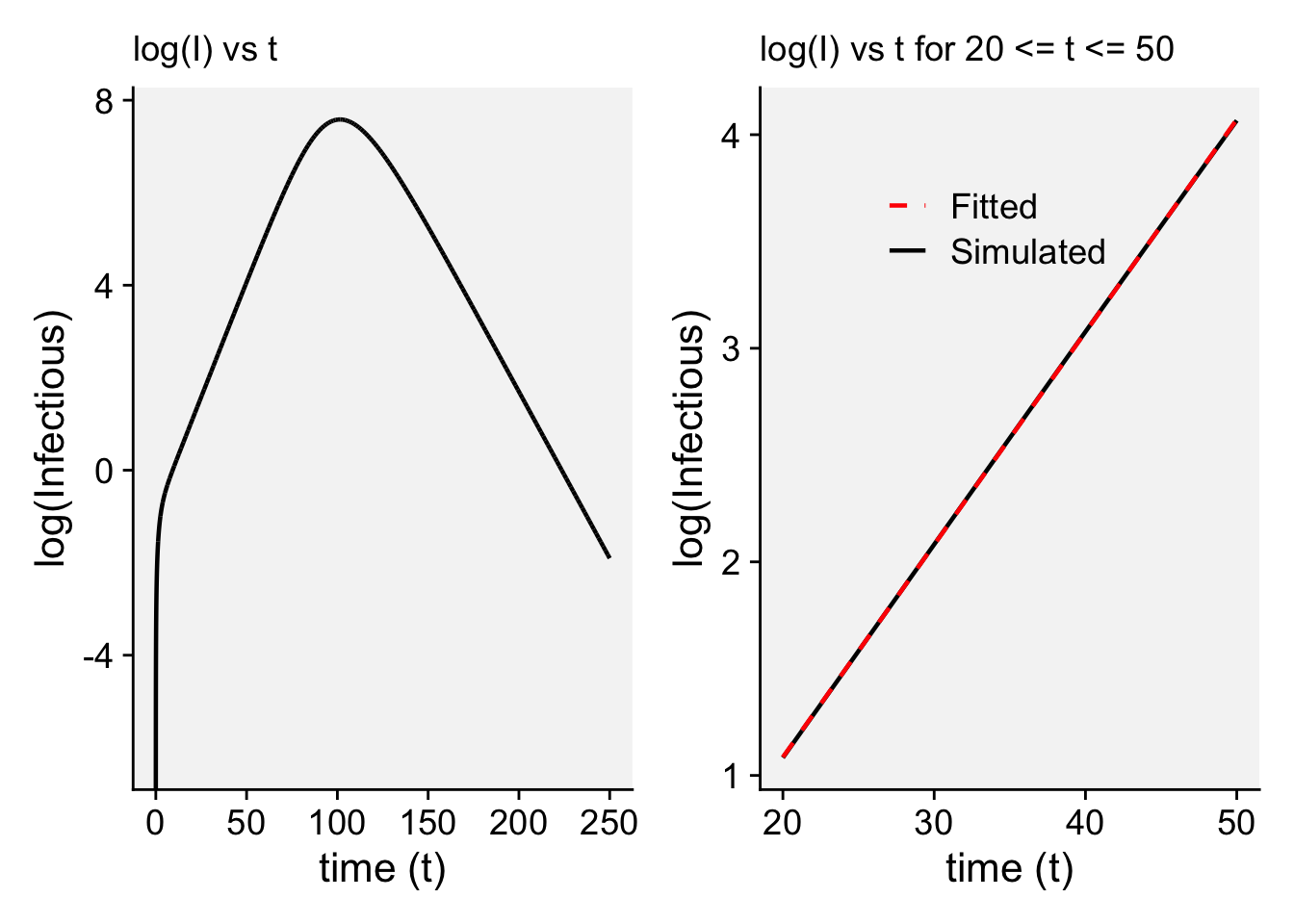
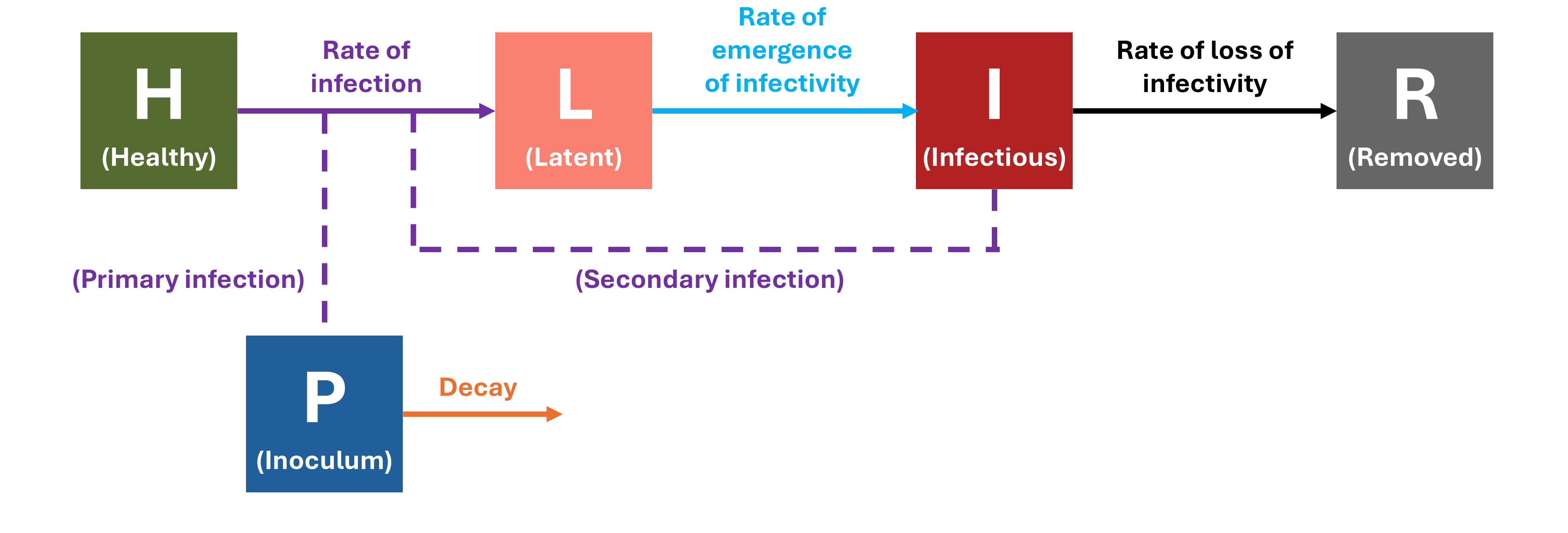
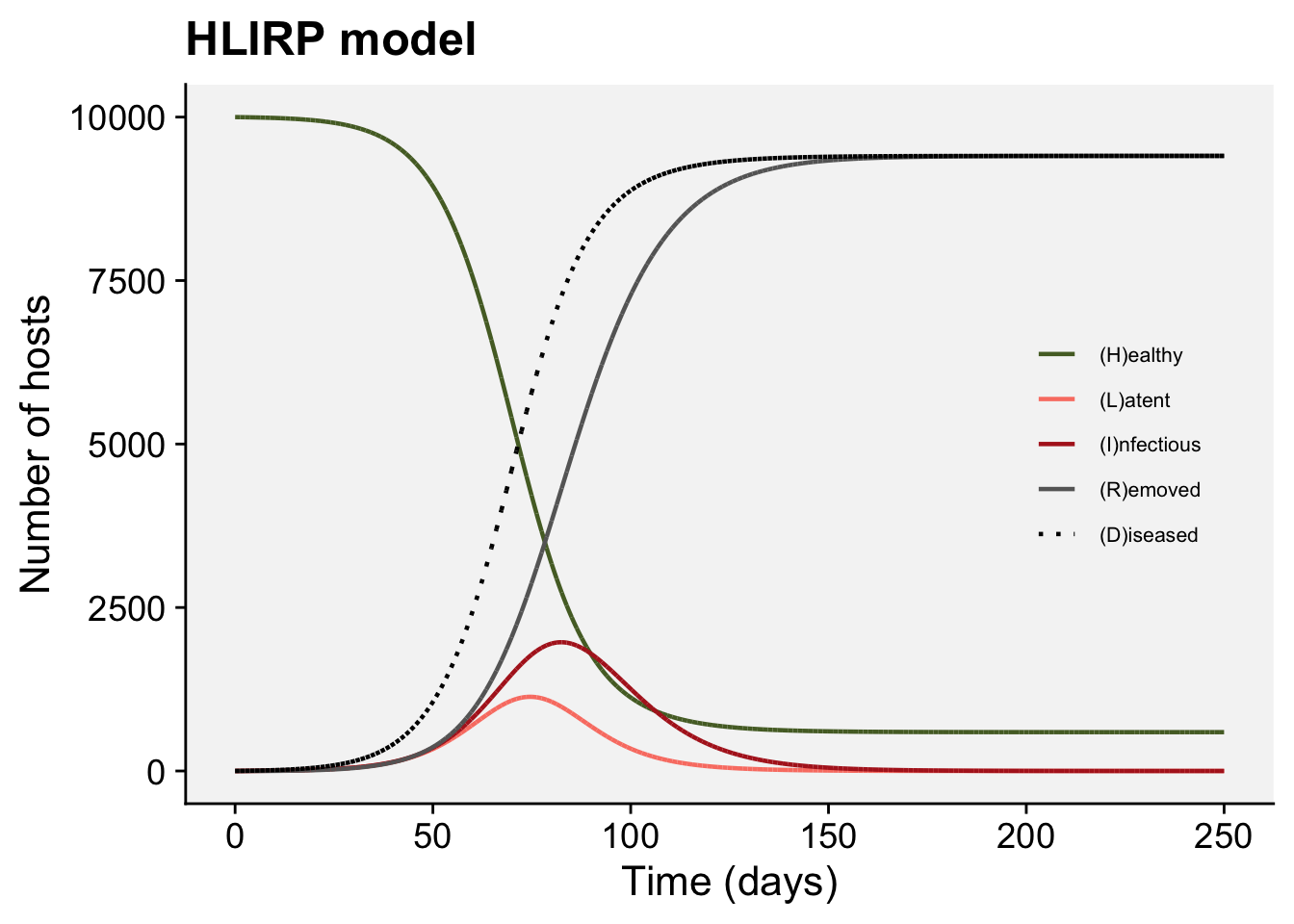
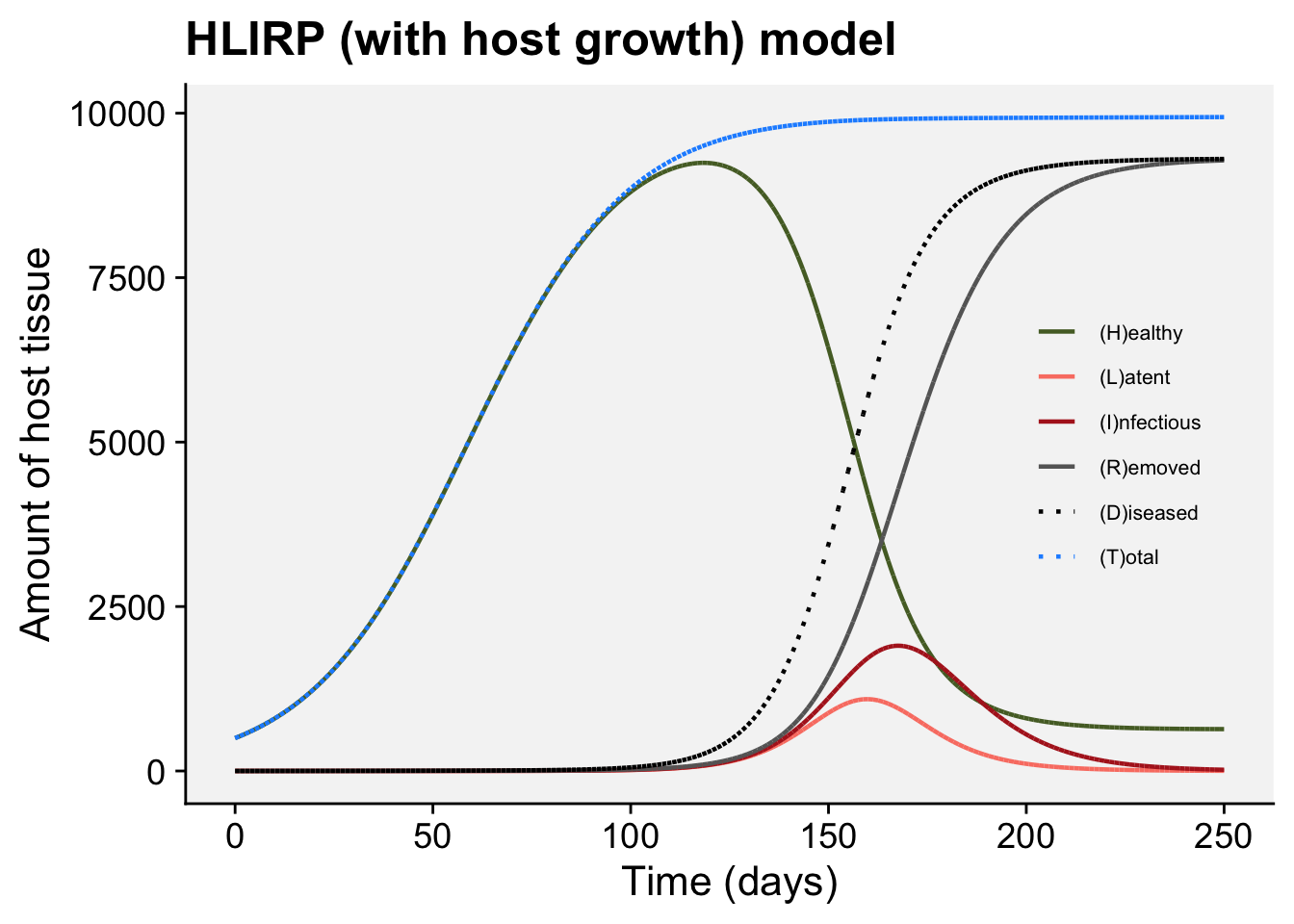
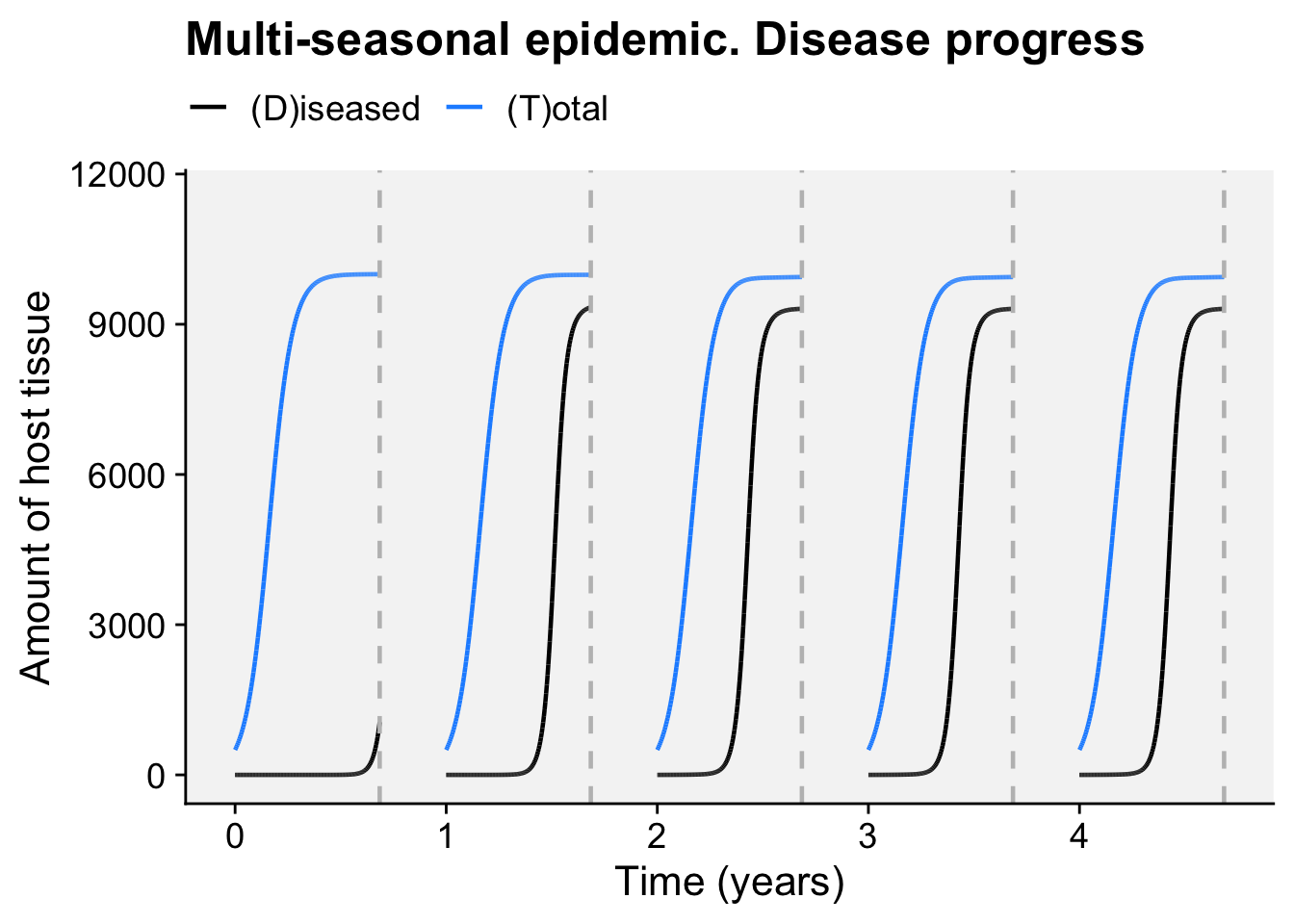
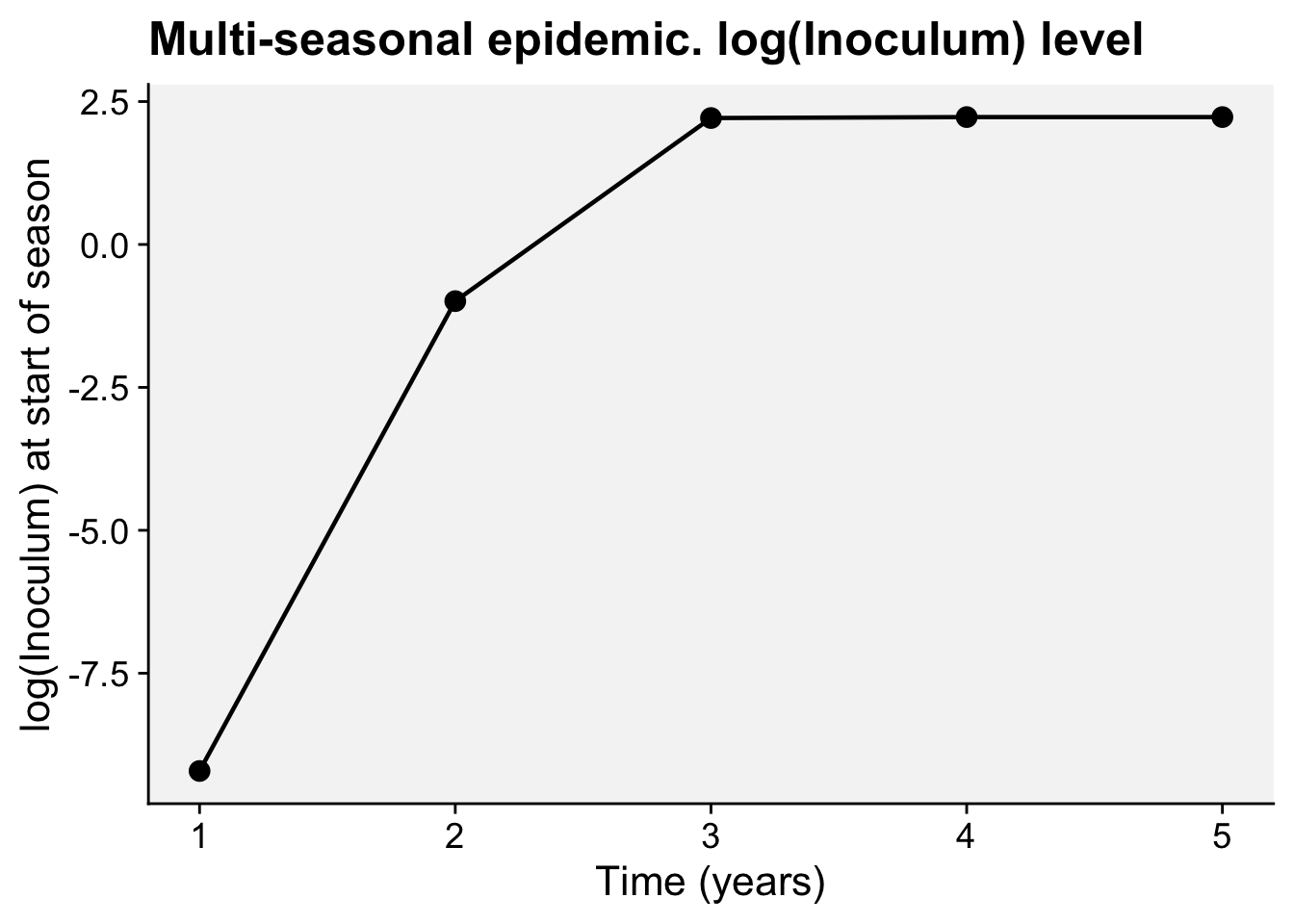
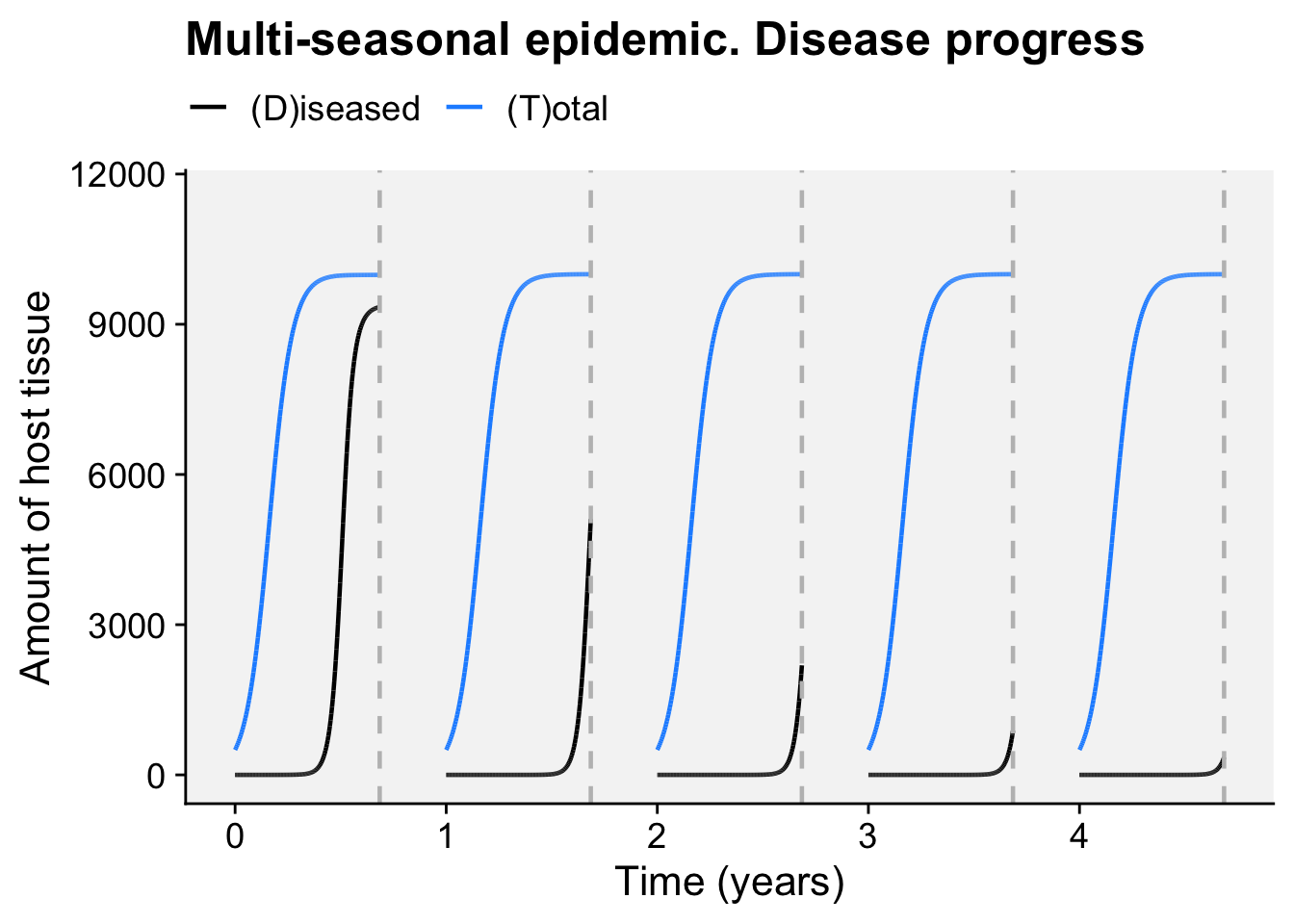
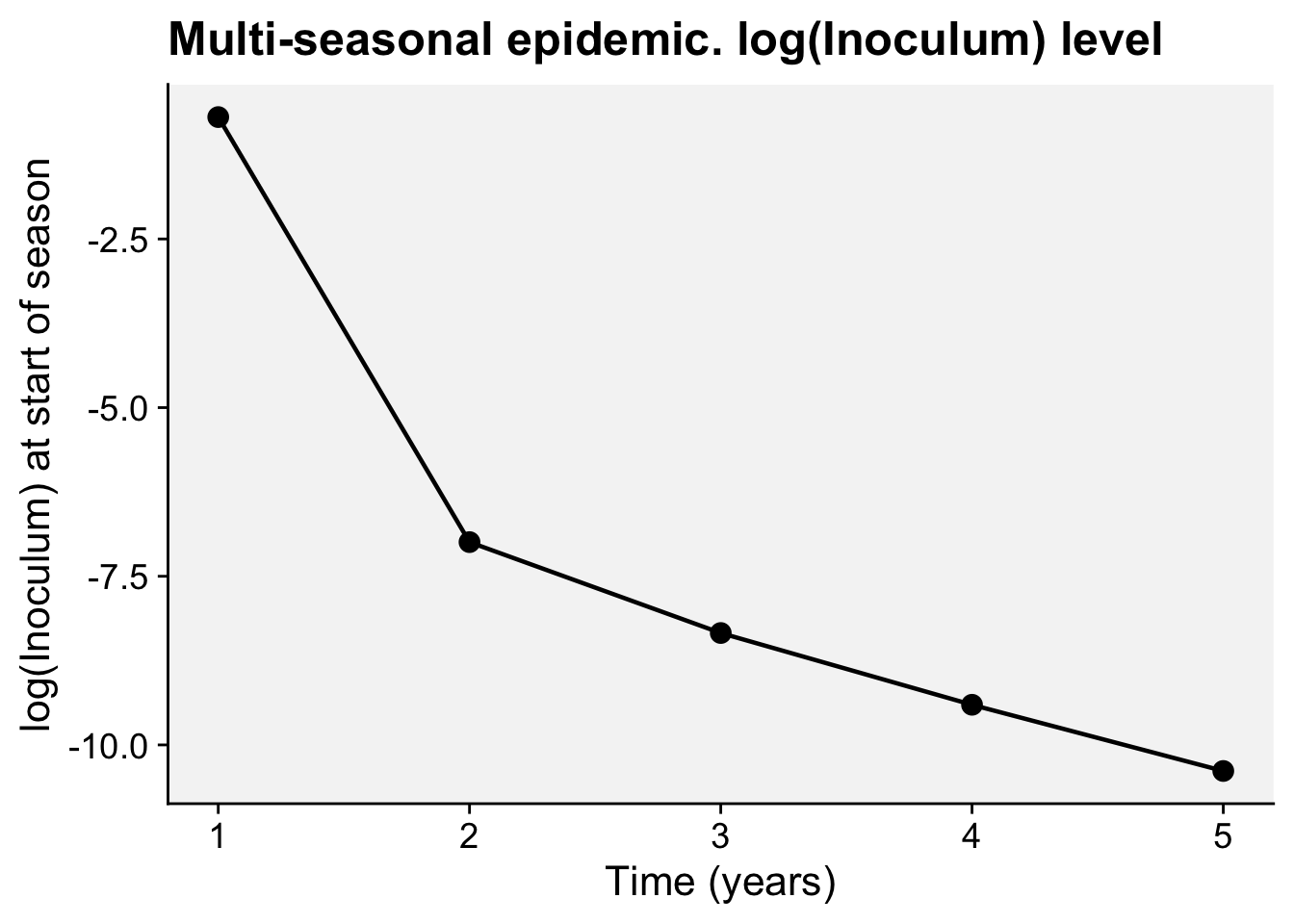
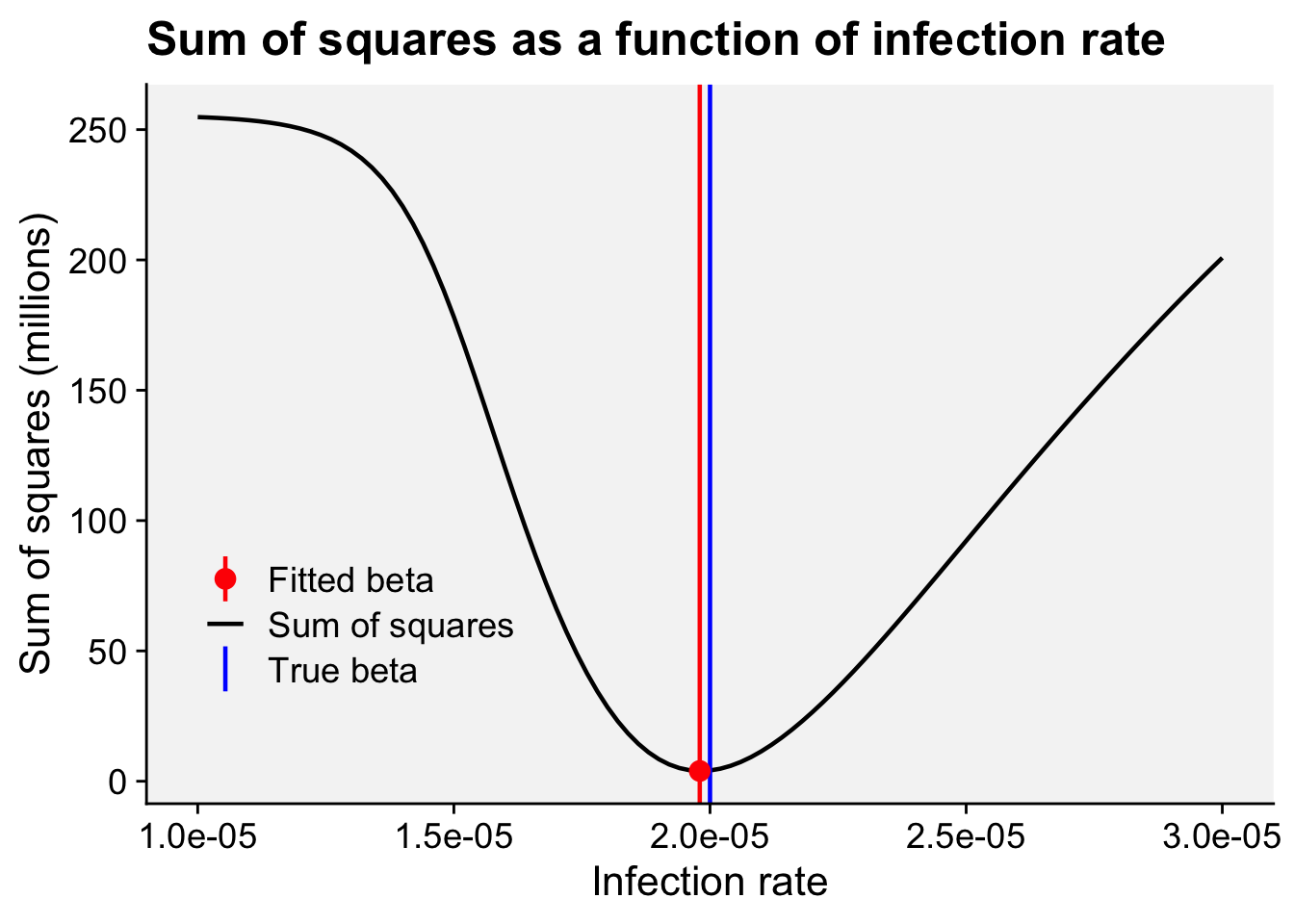
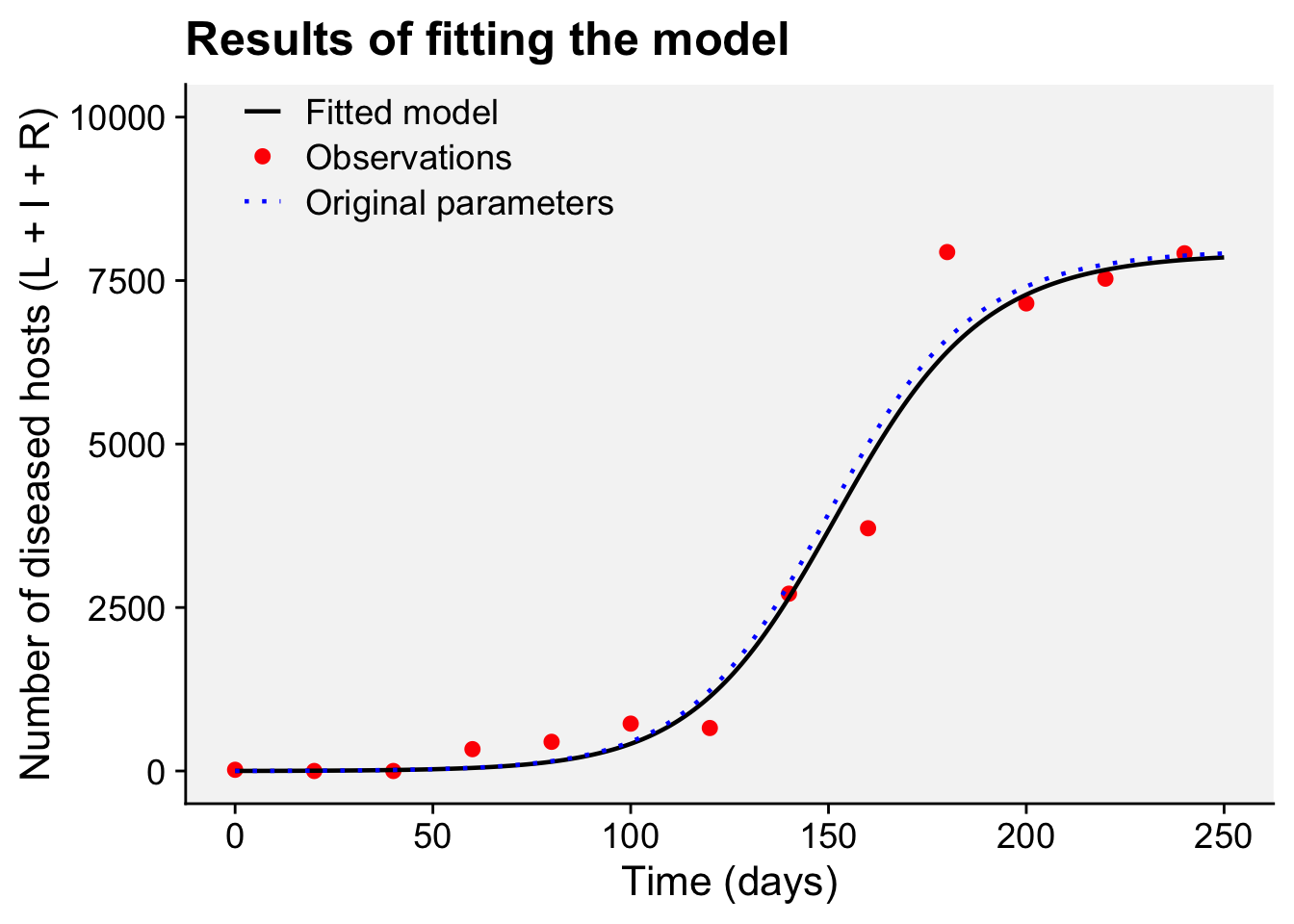
A concordance between process-based models and the Gompertz model can be made if we move to a more complex Kermack-McMcKermack type model in which the infection rate depends on the time-since-infection (Cunniffe et al. 2012; Segarra et al. 2001), at least approximately. However, this is really quite advanced and requires going well beyond what can be covered in this chapter, so we do not pursue this here.
Adrakey, H. K., Streftaris, G., Cunniffe, N. J., Gottwald, T. R., Gilligan, C. A., and Gibson, G. J. 2017. Controlling spatio-temporal epidemics using latent processes in a bayesian framework. Journal of the Royal Society: Interface 14:20170386.
Allen, L. J. S., Bokil, V. A., Cunniffe, N. J., Hamelin, F. M., Hilker, F. M., and Jeger, M. J. 2019. Modelling vector transmission and epidemiology of co-infecting plant viruses. Viruses 11:1153.
Best, A., and Cunniffe, N. J. 2026. Fitting a lattice model with local and global transmission to spread of a plant disease. PLOS Computational Biology 22:e1013404.
Brasset, P. R., and Gilligan, C. A. 1988. A model for primary and secondary infection in botanical epidemics. Journal of Plant Diseases and Protection 95:352–360.
Bussell, E. H., and Cunniffe, N. J. 2022. Optimal strategies to protect a sub-population at risk due to an established epidemic. Journal of the Royal Society: Interface 19:20210718.
Cunniffe, N. J., Cobb, R. C., Meentemeyer, R. K., Rizzo, D. R., and Gilligan, C. A. 2016. Modeling when, where and how to manage a forest epidemic, motivated by sudden oak death in california. Proceedings of the National Academy of Sciences 113:5640–5645.
Cunniffe, N. J., and Gilligan, C. A. 2020. Use of mathematical models to predict epidemics and to optimize disease detection and management. In Emerging plant diseases and global food security, eds. J. B. Ristaino and A. Records. American Phytopathological Society Press, pp. 239–266.
Cunniffe, N. J., Hamelin, F., Iggidr, A., Rapaport, A., and Sallet, G. 2024. Identifiability and observability in epidemiological models. A primer. Springer.
Cunniffe, N. J., Koskella, B., Metcalf, C. J. E., Parnell, S., Gottwald, T. R., and Gilligan, C. A. 2015a. Thirteen challenges in modelling plant disease. Epidemics 10:6–10.
Cunniffe, N. J., Stutt, R. J. O. H., Bosch, F. van den, and Gilligan, C. A. 2012. Time-dependent infectivity and flexible latent and infectious periods in compartmental models of plant disease. Phytopathology 102:365–380.
Cunniffe, N. J., Stutt, R. O. J. H., DeSimone, R. E., Gottwald, T. R., and Gilligan, C. A. 2015b. Optimising and communicating options for the control of invasive plant disease when there is epidemiological uncertainty. PLOS Computational Biology 11:e1004211.
Cunniffe, N. J., Taylor, N. P., Hamelin, F. M., and Jeger, M. J. 2021. Epidemiological and ecological consequences of virus manipulation of host and vector in plant virus transmission. PLOS Computational Biology 17:e1009759.
Del Ponte, E. M., Mikaberidze, A., Bebber, D. P., Halliday, F. W., and Cunniffe, N. J. 2026. The epidemiology of wild-crop interfaces: Integrating ecology, evolution and management through modelling. Philosophical Transactions B 381:20250110.
Donnelly, R., Cunniffe, N. J., Carr, J. P., and Gilligan, C. A. 2019. Pathogenic modification of plants enhances long-distance dispersal of non-persistently transmitted viruses to new hosts. Ecology 100:e02725.
Elderfield, J. A. D., Lopez-Ruiz, F. J., Bosch, F. van den, and Cunniffe, N. J. 2018. Using epidemiological principles to explain fungicide resistance management tactics: Why do mixtures outperform alternations? Phytopathology 108:803–817.
Ellis, J., Lazaro, E., Duarte, B., Magalhaes, T., Duarte, A., Benhadi-Marin, J., Pereira, J. A., Vicent, A., Parnell, S., and Cunniffe, N. J. 2025. Developing epidemiological preparedness for a plant disease invasion: Modelling citrus huánglóngbìng in the european union. Plants, People, Planet 7:1403–1423.
Fabre, F., Coville, J., and Cunniffe, N. J. 2021. Optimising reactive disease management using spatially explicit models at the landscape scale. In Plant diseases and food security in the 21st century, eds. P. Scott, R. Strange, L. Korsten, and L. Gullino. Springer.
Fabre, F., Rousseau, E., Mailleret, L., and Moury, B. 2012. Durable strategies to deploy plant resistance in agricultural landscapes. New Phytologist 193:1064–1075.
Falla, E. K., and Cunniffe, N. J. 2026. The benefits of an attractive companion: Modelling control of non-persistently transmitted plant viruses via trap or repellent companion plants. Plant Pathology 75:e70171.
Gibson, G. J., and Austin, E. J. 1996. Fitting and testing spatio-temporal stochastic models with application in plant epidemiology. Plant Pathology 45:172–184.
Gubbins, S., and Gilligan, C. A. 1997. Biological control in a disturbed environment. Philosophical Transactions of the Royal Society of London, B. 352:1935–1949.
Hamelin, F. M., Allen, L. J. S., Bokil, V. A., Gross, L. J., Hilker, F. M., Jeger, M. J., Manore, C. A., Power, A. G., Rúa, M. A., and Cunniffe, N. J. 2019. Coinfections by noninteracting pathogens are not independent and require new tests of interaction. PLOS Biology 17:e300551.
Hamelin, F. M., Castel, M., Poggi, S., Andrivon, D., and Mailleret, L. 2011. Seasonality and the evolutionary divergence of plant parasites. Ecology 92:2159–2166.
Hyatt-Twynam, S. R., Parnell, S., Stutt, R. O. J. H., Gottwald, T. R., Gilligan, C. A., and Cunniffe, N. J. 2017. Risk-based management of invading plant disease. New Phytologist 214:1317–1329.
Keeling, M. J., and Rohani, P. 2008. Modeling infectious diseases in humans and animals. Princeton University Press.
Madden, L. V. 2025. Reflections on the past, present, and future of quantitative plant disease epidemiology. Annual Review of Phytopathology 63:1–22.
Madden, L. V., Hughes, G., and van den Bosch, F. 2007.
The study of plant disease epidemics. The American Phytopathological Society.
https://doi.org/10.1094/9780890545058.
Madden, L. V., and van den Bosch, F. 2002. A population-dynamics approach to assess the threat of plant pathogens as biological weapons against annual crops. BioScience 52:65–74.
Murray-Watson, R. E., and Cunniffe, N. J. 2023. Expanding growers’ choice of disease management options can promote suboptimal social outcomes. Plant Pathology 72:933–950.
Russell, R., and Cunniffe, N. J. 2025. Optimal control prevents itself from eradicating stochastic disease epidemics. PLOS Computational Biology 21:e1012781.
Segarra, J., Jeger, M. J., and van den Bosch, F. 2001. Epidemic dynamics and patterns of plant disease. Phytopathology 91:1001–1010.
Taylor, N. P., and Cunniffe, N. J. 2023. Optimal resistance management for mixtures of high-risk fungicides: Robustness to the initial frequency of resistance and pathogen sexual reproduction. Phytopathology 113:55–69.
Thompson, R. N., Gilligan, C. A., and Cunniffe, N. J. 2018. Control fast or control smart: When should invading pathogens be controlled? PLOS Computational Biology 14:e1006014.
Thompson, R. N., Gilligan, C. A., and Cunniffe, N. J. 2020. Will an outbreak exceed available resources for control? Estimating the risk from invading pathogens using practical definitions of a severe epidemic. Journal of the Royal Society: Interface 17:20200690.
van der Plank, J. E. 1963. Plant diseases: Epidemics and control. Academic Press.
Vincent, E. M. R., Hill, E. M., and Parnell, S. 2025. Modelling the effectiveness of integrated pest management strategies for the control of septoria tritici blotch. PLOS Computational Biology 21:e1013352.
Willocquet, L., Savary, S., McDonald, B. A., and Mikaberidze, A. 2020. A polyetic modelling framework for plant disease emergence. Plant Pathology 69:1630–1643.
Zadoks, J. C. 1974. The role of epidemiology in modern phytopathology. Phytopathology 64:918–923.
Zaffaroni, M., Rimbaud, L., Mailleret, L., Cunniffe, N. J., and Bevacqua, D. 2021. Modelling interference between vectors of non-persistently transmitted plant viruses to identify effective control strategies. PLOS Computational Biology 17:e1009727.